
Abstract
Modern biological dependency on trace elements is proposed to be a consequence of their enrichment in the habitats of early life together with Earth’s evolving physicochemical conditions; the resulting metallic biological complement is termed the metallome. Herein, we detail a protocol for describing metallomes in deep time, with applications to the earliest fossil record. Our approach extends the metallome record by more than 3 Ga and provides a novel, non-destructive method of estimating biogenicity in the absence of cellular preservation. Using microbeam particle-induced X-ray emission (µPIXE), we spatially quantify transition metals and metalloids within organic material from 3.33 billion-year-old cherts of the Barberton greenstone belt, and demonstrate that elements key to anaerobic prokaryotic molecular nanomachines, including Fe, V, Ni, As and Co, are enriched within carbonaceous material. Moreover, Mo and Zn, likely incorporated into enzymes only after the Great Oxygenation Event, are either absent or present at concentrations below the limit of detection of µPIXE, suggesting minor biological utilisation in this environmental setting. Scanning and transmission electron microscopy demonstrates that metal enrichments do not arise from accumulation in nanomineral phases and thus unambiguously reflect the primary composition of the carbonaceous material. This carbonaceous material also has δ13C between −41.3‰ and 0.03‰, dominantly −21.0‰ to −11.5‰, consistent with biological fractionation and mostly within a restricted range inconsistent with abiotic processes. Considering spatially quantified trace metal enrichments and negative δ13C fractionations together, we propose that, although lacking cellular preservation, this organic material has biological origins and, moreover, that its precursor metabolism may be estimated from the fossilised “palaeo-metallome”. Enriched Fe, V, Ni and Co, together with petrographic context, suggests that this kerogen reflects the remnants of a lithotrophic or organotrophic consortium cycling methane or nitrogen. Palaeo-metallome compositions could be used to deduce the metabolic networks of Earth’s earliest ecosystems and, potentially, as a biosignature for evaluating the origin of preserved organic materials found on Mars.
Similar content being viewed by others

Introduction
“The system of cell chemistry…cannot be divorced from the environment any more than it can be separated from a code. All the basic chemicals and energy come from the environment and this remains true to this day”
R. J. P. Williams and J. J. R Fraústo da Silva (Journal of Theoretical Biology, 2003).
A wide range of carbonaceous materials (CM) occur in Palaeoarchaean (~3.6-3.2 Ga) rocks. Some CM is demonstrably biogenic1,2, however, due to maturation, metamorphism and the difficulty inherent in distinguishing between CM derived from biogenic and abiotic processes, the origin of most Archaean CM remains controversial3,4. This is further compounded by the fact that both biogenic and abiotic CM would have been generated in early habitable environments, for example, hydrothermally influenced oceans with significant exogenous input1,4,5,6. As a result, the fossil record of early life, and especially the potential fossil record encoded in Palaeoarchaean CM, remains incompletely understood3,7. Nonetheless, similarities in the occurrence, morphology and chemistry of CM from similar settings should support common origins. Generally, abiotic processes are more likely to form CM with an unexceptional continuum of geological-biogeochemical characteristics, whereas more striking patterns with restricted ranges in chemical characteristics are expected in biogenic CM in spite of subsequent alteration2,8,9,10. Such patterns, when discerned in Archaean CM, usually have the hallmarks of metamorphosed biological material in terms of C-N-S isotope geochemistry, spectral characteristics from, e.g., Raman and IR spectroscopy, and the spatial correlation of CHNOPS elements, albeit muted compared to bio-indicative signals in extant biomass2,5,8,10,11. Quantitative approaches to characterising biogeochemistry in deep time are therefore of great value in ameliorating our understanding of Precambrian palaeoecology. In the case of unambiguously biogenic CM, for example microbial mat laminations, repeated similarities in isotopic, spectral or molecular characteristics might indicate the metabolic affinities of precursor biomass (e.g.9,10,12,13). Regrettably, although carbon isotope ratios related to microfossils and organo-sedimentary structures can be interpreted as the effects of organisms or consortia9,12,14, δ13C values mostly between −40‰ and −10‰ reported for Archaean “biomass” are consistent with multiple biological pathways, for example, anoxygenic photosynthesis, sulphate-reduction, photoferrotrophy and methanogenesis15,16,17. Furthermore, these values fall within the range exhibited by the products of abiotic Fischer-Tropsch-type processes (e.g.18), which were widespread in the environments of early life.
We here further the range of biogeochemical proxies for decoding palaeoecology by proposing that CM of otherwise enigmatic origins (Fig. 1) could, when preserving specific trace element enrichments, be diagnostic of the metabolism of precursor biomass. We term this trace element signature the “palaeo-metallome”, in recognition of the fact that it represents a remnant of chemical selectivity by cells leading to their inorganic elemental composition (the “metallome”)19,20. Metals facilitate numerous biosynthetic processes as parts of oxidoreductases and other metalloenzymatic and proteinic nanomachines, in addition to being bound by extracellular polymers (e.g.20,21,22,23). Decoding patterns of metal enrichment in fossil biomass, historically difficult due to limits of instrumental resolution and limited understanding of the rapidity of silicification processes, may be achieved using quantified ion-beam analyses at high resolution. Although microbial material is capable of metal retention even after cell death22,24,25, early and post-diagenetic effects in rocks with poor preservation potential may alter elemental compositions via leaching and fluid remobilisation22,24. Very rapidly silicified, exceptionally preserved CM that may be able to preserve relic bio-accumulation of trace metals without post-diagenetic alteration is, however, common in Archaean chert horizons1,4,5.

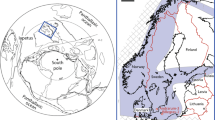
Sampling location maps (a,b) and petrographic context of the microstructures of interest; irregular clots (c,d) and volcanic particle coatings (e,f) sourced from the 3.33 Ga Josefsdal Chert (Barberton greenstone belt, South Africa). (a) Map of southern Africa showing the location of the Barberton greenstone belt. (b) Map of the southern portion of the Barberton greenstone belt; white box indicates the location of the Josefsdal Chert, near the border of South Africa and Eswatini, from which all studied samples were collected. (c) Thin section of weakly laminated clotted carbonaceous chert corresponding to black massive chemical chert observed in the field (see Fig. S2). Carbonaceous clots are distributed throughout a silica matrix; other phases include scattered microminerals including pyrite and chromite spinel (see Fig. 2). (d) High-resolution photomicrograph showing irregular, stellate carbonaceous clots of up to ~1 mm diameter. (e) Thin section of laminated black and white banded chert corresponding to laminated chert observed in the field (see Fig. S2). (f) High-resolution photomicrograph showing sub-rounded and sub-angular pseudomorphed and silicified volcanic particles coated and impregnated with carbonaceous material (dark regions).
Rationale: A Framework for Estimating the Palaeo-Metallome of Fossil Biomass
The metallome is the component of biosynthetic chemistry comprising the metals and metalloids of a biological system, i.e., the inorganic complement to the proteome and genome19,20,21,26. In palaeobiological terms, the metallome is best appraised as the totality of elements within cellularly derived fossils. Currently, the oldest metallomes date from 131-120 Ma feathers27 and 50 Ma leaves28, but it has nonetheless been suggested that linking metal signatures from fossils in deep time to their organic chemistry may illuminate metallome evolution throughout geological history29,30. The relative proportions of elements in well-preserved CM may also be linked to their presence in proteins, metabolites and other biomolecules within the predicted biological system26. A palaeo-metallomic biosignature in the rock record should therefore chart systematic, bioaccumulative interactions between organisms and the external environmental compartment sourcing the elements that constitute the metallome21.
If modern biological dependency upon trace metals is a consequence of the richness of these elements in the environments of early life20,21, proposing a palaeo-metallomic signature in deep time demands a framework of palaeoenvironmental reconstruction. The 3.33 Ga Josefsdal Chert (JC; equivalent to Kromberg Formation unit K3c, Barberton greenstone belt; Figs. 1a,b, S1,S2), from which our samples were collected, is best represented by outcrops between Ekulindeni, Mpumalanga province, South Africa, and Bulembu, Hhohho region, Eswatini. The JC is an archetypal Palaeoarchaean chert comprising silicified volcanogenic sediments intercalated with near-pure chert horizons and chemical sediments. These horizons contain variably preserved microbial vestiges in the forms of microfossils and microbial mats31,32. The three-dimensional preservation of such biosignatures5 stands testament to their rapid silicification, which necessarily commenced during the life cycle of the organism33. Moreover, the JC presents copious outcrop and geochemical evidence for widespread hydrothermally influenced sedimentary fabrics31,32; Fig. S2). The organisms inhabiting such environments are estimated to have been (hyper)thermophilic and, if immediately downstream of hydrothermal effusions, likely chemosynthetic with polyextremotolerance to salinity and biotoxic metal enrichments14,31,33,34,35. Regrettably, the chemosynthetic biosphere leaves enigmatic vestiges of its existence in the rock record, beset by simple morphologies and subsequent alteration by diagenetic and metamorphic processes even where rapidly preserved by silicification14,31,33. The Palaeoarchaean fossil record nonetheless presents an ideal window on the primitive thermophilic biosphere: it dates from a time when life on Earth was driven dominantly by internal heat35,36,37,38,39. Both chemotrophic and phototrophic metabolisms have been predicted as early as 3.8 Ga11,12,15,35,40 and the probability of chemosynthetic inhabitants in the palaeoenvironments represented by Archaean cherts makes these rocks ideal text cases for palaeo-metallomics, since such extremotolerant thermophiles have unique metallome compositions with specific elemental requirements41.
Following palaeoenvironmental reconstruction, deducing palaeo-metallomic signatures demands petrographic and geochemical demonstration that the CM studied is of biogenic origin. This entails proof of CM syngenicity, ideally using high-magnification optical petrography coupled to Raman spectroscopy and geothermometry, and carbon isotope geochemistry to indicate negative δ13C values consistent with biological fractionation.
Deducing metallomic biosignatures in Precambrian rocks (the main focus of this study) demands the detection of metal and metalloid signatures within CM, and will pose very different challenges to the determination of metallomes in modern and sub-fossil samples. Although it should be possible to calculate the elemental composition of CM when present in sufficiently high concentrations, linking elemental concentrations to specific functions within the genome or proteome is impossible due to the lack of preservation of biomolecules from the genome or proteome in ancient fossils27,42. We propose that it is more prudent to consider a conservative approach akin to that of Phanerozoic metallome studies involving the quantification of the spatial distribution of trace metals within primary structures of plausible biological origin followed by an attempt to logically link these metal enrichments to biosynthetic pathways possible within the constraints of the palaeoenvironment26,27,28. Furthermore, ancient samples, particularly often-controversial carbonaceous materials in Archaean cherts, require proof that post-diagenetic processes did not contribute to the eventual signal22 or, where present, that such overprints can be quantified.
Results: Testing the Palaeo-Metallome Hypothesis
Step I: Palaeoenvironmental reconstruction
The “time capsule” preservation of morphological and geochemical characteristics afforded by silicification, i.e. rapid encapsulation, lithification, and close to zero post-diagenetic alteration, is a unique taphonomic setting43. The extremely rapid silicification characterising Archaean cherts results from a combination of the high (oversaturated-supersaturated) Si content of the Archaean oceans due to both dissolution of seafloor volcanics and the lack of biological uptake of silica into tests and shells with respect to the modern, coupled with Si-rich hydrothermal effluence, which was demonstrably more significant on the Archaean Earth than at present31,34. The resulting rapidity of preservation assures that pristine and unaltered geochemical signatures are retained (Fig. 2), for example REE+Y compositions defining palaeodepositional conditions (Fig. 2a), Raman spectral characteristics demonstrating that the thermal maturity of this disordered CM is commensurate with that of the host rock (Figs. 2b–d, S3), minerals and materials resistant to silicification (Fig. 2e–h) and isotopic compositions evidencing fractionation pathways in the precursor materials (Fig. 3).

Selected geochemical analyses relevant to the palaeoenvironmental characterisation of the 3.33 Ga Josefsdal Chert. (a) REE+Y compositions of various samples from the studied horizons of the Josefsdal Chert normalised to Mud from Queensland (MuQ), a mixed mafic-felsic shale estimate of Archaean continental composition. HREE enrichment and positive La and Y anomalies indicates hydrogenous, thalassogenic contributions (marine), whereas positive Eu anomalies indicate hydrothermal fluid contributions. (b,c) Raman spectroscopy mapping of a representative irregular clot (b) and carbon-impregnated volcanic particle (c). Inset optical images show the regions of analysis; field of view = 1 mm. Green = carbonaceous material; yellow-orange = quartz; blue = anatase. Anatase can be considered a proxy for the alteration of volcanic particles associated with carbonaceous material. (d) Average Raman spectra for carbonaceous material in four studied samples (99SA07, black; 12SA09, green; 12SA16, blue; 14SA01, red). (e,f) SEM-EDS analyses of non-silicified mineral phases associated with carbonaceous material. Spectra are colour-correspondent to points within the regions of analysis shown. Red spectra = Ni-rich pyrite; beige spectra = K-Al phyllosilicate. Extensive SEM-EDS analyses are shown in Figs. S4-S8. (g,h) TEM micrographs from within clotted microstructures (sample 99SA07) showing the discontinuous distribution of carbonaceous material (black) within the microquartz matrix (grey).

Box-plot representations of SIMS δ13C data for irregular clots, particle coatings/impregnations and generic carbonaceous material (other CM) in the studied samples of the Josefsdal Chert. Comparisons are given with well-defined carbon isotope fractionations for the products of anoxygenic and oxygenic photosynthesisers, and methane and sulphur-cycling organisms (compiled after McCandless and Gurney77; Schidlowski15; Londry and Des Marais76; Vieth and Wilkes16). Well-defined microstructures (clots and coatings) have negative and restricted δ13C ranges consistent with biological mediation by multiple pathways. Coatings are characterised by δ13C values slightly more negative than clots (averaging −15.16‰ versus −9.44‰). Generic CM (other CM) in the same samples is characterised by a wide range of positive and negative δ13C values (between −47.4 ± 8.3‰ and +13.5 ± 2.0‰) bearing less resemblance to products resulting from specific biological pathways.
Normalised REE+Y patterns (measured using ICP-MS) are characterised by fractionated trends with weak HREE enrichment ((Pr/Yb)MUQ = 0.57 to 2.41), mostly positive La anomalies [La/La*MUQ = LaMUQ/(Pr*MUQ(PrMUQ/NdMUQ)2)] between 0.93 and 2.90, the absence of negative Ce anomalies, positive Eu anomalies [Eu/Eu*MUQ = EuMUQ/(Sm2MUQ * TbMUQ)1/3] between 1.01 and 2.02, and positive Y anomalies [Y/Y*MUQ = YMUQ/(0.5ErMUQ * 0.5HoMUQ)] between 1.13 and 1.45 (i.e., superchondritic Y/Ho ratios between 30.31 and 38.57). La, Y, Y/Ho and Eu systematics indicate that the palaeodepositional setting of these cherts was influenced by marine and hydrothermal fluids44, with muted anomaly characteristics relative to pure marine precipitates indicating basin restriction45 and increased terrigenous influence46, likely from exposed mafic-felsic landmasses. High concentrations of Fe- and potentially Mn-oxyhydroxides in semi-restricted disequilibrium settings such as this would have stimulated the concentration of trace and rare earth elements through their induced dissolution, enhancing the local bioavailability of these elements44,46. The environment of the Josefsdal Chert was therefore metal-rich, warm-hot, anoxic, and replete with chemical disequilibria as a result of the confluence of discrete fluid reservoirs. The studied samples thus reflect a habitable environment that seems to have been typical on the early Earth (see Discussion of38).
Step II: Syngenicity and biogenicity of CM
Two petrographically distinct morphologies of CM were studied using secondary ion mass spectrometry and microbeam particle-induced X-ray emission (µPIXE): i) irregular, stellate clots of 200–2,000 µm diameter (Fig. 1c,d) and ii) non-isopachous coatings on altered volcanic particles (Fig. 1e,f). Both of these microstructures occur within the same millimetre-scale domains as the well-preserved microbial mats and biofilms in the JC, i.e., within the black bands of black-and-white banded cherts and within carbon-bearing regions of massive black cherts (Figs. 1, S2). Microbial mats either drape over clotted textures or coexist with them in the form of incompletely formed biofilm fragments. The studied clots are ‘free-floating’ microstructures in volcano-hydrothermal sediments, co-occurring with degraded microbial mats, and were interpreted as putative degraded chemosynthetic biomass growing in situ within a gel-like sediment by Westall et al.31. Archaean clotted carbonaceous material was first ascribed a potential biological origin by Walsh47, who compared it to microbe-mineral aggregates in modern marine environments48,49. Coating-like fabrics on volcanic particles were interpreted as putative lithotrophic coverings of specific volcanogenic fragments based on similarity to known microfossils (cf.5,31). Neither case for biogenicity has yet been unambiguously demonstrated, however, there are many very strong arguments against these CM morphologies being abiotic in origin, the full reasoning for which is explained in the Supplementary Material. In brief: (i) non-isopachous, irregular morphologies argue against an origin as condensation fabrics seen in contemporaneous CM50; (ii) the lack of continuous, blanket-like carbonaceous laminae with grain-supported textures argues against an origin as either meteoritic6 or detrital, sequestered CM (see13); (iii) no aggregates with radioactive cores have been identified, therefore an origin as pyrobitumen51 is not possible; (iv) no CM-infused tephra-like morphologies52 have been observed; and (v) the restricted range of δ13C is inconsistent with FTT-derived hydrocarbons18,53. Thus, no plausible abiological interpretation for either the irregular clot or particle coating CM morphologies is sustained under scrutiny. Conversely, a biological origin is strongly supported by palaeoenvironmental and petrological context31,32 (Fig. 1), for example that laminated and clotted CM alike would have been able to grow through attachment on, or matrix-supported suspension within, a gel-like Si-rich volcanogenic/chemical sediment. Although previous studies e.g.1,47 have inferred that there may be a genetic link between mats and clots, there is no evidence for this in our samples. Specifically, since mats and clots occur within the same horizons without evidence for differential compression, the two must have developed simultaneously.
Raman spectra are qualitatively characteristic of disordered carbon with a broad D band at 1,350 cm−1 and minor G band at 1,580 cm−1. Coupled with peak metamorphic temperatures of between 285 °C and 298 °C estimated according to the Raman geothermometer of Kouketsu et al.54, these spectra indicate that all studied CM has a thermal history consistent with that of the host rock (Fig. 2b–d), i.e., the CM is syngenetic. The studied microstructures have δ13C compositions between −41.3‰ and +0.03‰, averaging −9.44‰ and −15.16‰ for clots and coatings, respectively (Fig. 3; Table S1), consistent with biological fractionation by multiple pathways as reported in contemporaneous rocks (e.g.9,15,16,17 and references therein). δ13C values are slightly more negative in coatings than clots. Both microstructures are characterised by a restricted range of δ13C values compared to generic carbonaceous fragments and particles of uncertain origins (i.e., abiotic or biotic) in the same samples (Fig. 3; Table S1). Since the biogenicity of these microstructures is probable, it becomes justifiable to calculate the trace element composition of this CM in the framework of the palaeo-metallome.
Step III: Trace element signatures in CM
µPIXE was performed at the microbeam beamline of the AIFIRA facility described in Sorieul et al.55. Multiple examples of each CM morphology were measured. Their adjacent matrices i.e., a relic of the palaeodepositional fluid, were used as the ambient concentration against which absolute elemental concentrations within specific microstructures were compared. Multiple analyses of the matrix were conducted in each sample, providing average matrix values for elemental concentration that were found to be indistinguishable from bulk rock ICP-MS measurements (Figs. S10-S11; Table S3), i.e. ICP-MS and µPIXE results are, as expected, directly comparable. µPIXE was used to map elements in the mass range from P to beyond Mo with ppm accuracy56,57.
Excluding enrichments due to micromineral phases (which should not be taken as a part of the metallomic signature, since it is usually not possible to distinguish microbially mediated and abiotic precipitates from elemental fractionation and concentration alone58), we found that irregular clots exhibit significant (>150% relative to matrix concentrations) enrichments (e.g., Figs. 4a–c, S12) in nine bio-functional elements: P, S, Ti, V, Fe, Co, Ni and As. Furthermore, Mn, Cu, Zr and Mo appear modestly enriched (100–150%) relative to the matrix, although all of these and Zn occur at concentrations below the limit of detection appropriate for quantification (Table S2). Ca and Cr are depleted (<100%) relative to the matrix. Although P is depleted relative to the matrix in two analyses, high densities of apatite (Ca5(PO4)3) crystals were observed in the adjacent matrix, and P concentrations within CM are of the same order of magnitude as in analyses where CM is enriched. Average elemental concentrations in irregular clots follow the trend: K > Ti > Fe, P > V > Ni > Cr > As, S > Co > Cu > Zn > Zr, Mo, Mn. Elements listed in italics were mapped and are thus present but at concentrations below the theoretical limit of quantification. Anomalously high S concentrations in some analyses are attributable to micro-pyrite grains that are intimately associated with CM. Such morphologies and occurrences of pyrite are consistent with microbial metabolism, for example after degradation of biogenic organic material by sulphate-reducing bacteria or the anaerobic oxidation of methane (e.g.14,59,60). Although micro-pyrites identified in SEM imaging are point sources for As, Ni and Cu (≤11,500 ppm), these elements are also broadly and diffusely distributed throughout CM in irregular clots (Figs. 4, S12).

µPIXE spatially quantified elemental mapping within irregular clots. (a) µPIXE optical image of representative irregular clot. White box indicates the region for which the spectrum (b) and element maps (c) are given. (b) µPIXE spectrum with peak identification for the energy range 0–18 keV. (c) Element heat maps (black = low concentration, white = high concentration) for the region indicated in A, showing localised significant enrichments corresponding to entrained micromineral particles such as those identified by SEM-EDS in Fig. 2, but also a general enrichment within carbonaceous material (outlined by dotted white line) relative to the matrix.
Coatings of CM on volcanic particles show similar elemental enrichments to clots, but with inconsistent P distributions. Coatings are significantly enriched relative to the matrix in S, V, Ni, Cu, As and Zr, although Zn concentrations are below the limit of detection for quantification (Figs. 5a, S13). K, Fe and Co are modestly enriched, whereas P, Ca, Ti and Cr are depleted. Mn, Zn and Mo are present at concentrations below the limit of detection for quantification. Anomalously high Ti and Cu values in some analyses may results from microscopic ilmenite (into which Cu can substitute), titanomagnetite and anatase intermixed with volcanogenic material and CM (identified via SEM-EDS and Raman spectroscopy) or from accumulation associated with biotoxicity (after33). Average elemental concentrations in clots follow the trend: K, Ni > Cu, Fe > V > S > As > P, Ti > Co > Zr > Ca > Zn, Mo, Mn. In both irregular clots and particle coatings, a trend in decreasing absolute concentration of trace elements with atomic mass is observed, whereas enrichment relative to the matrix is element-dependent (Figs. 5a, S13, Table S2).

µPIXE spectra after mapping within carbon-rich volcanic particle coatings (a) and generic carbonaceous material in the form of flakes (b). (a) µPIXE optical image of representative particle coating and accompanying spectra with peak identification for the energy range 0–18 keV. (b) µPIXE optical image of a representative carbonaceous flake and accompanying spectra with peak identification for the energy range 0–18 keV. Note the very strong enrichments in certain transition metals within the flake (Fe, Ni, Ti) corresponding to their spatially restricted accumulation within micro-mineral particles as shown in Fig. S15-S16.
Generic flakes, particles and fragments of CM throughout rocks of the same cherts that do not conform to any specific microstructure (e.g., Figs. 5b, S14) were analysed as a comparison to the clot and coating microstructures. Although there is again a decreasing absolute enrichment with atomic mass (Fig. S15, Table S2), specific elemental enrichments are either minor or inexistent relative to the matrix (Fig. 5b). High-resolution µPIXE mapping shows that transition metal enrichments are invariably linked to microminerals in generic CM (Fig. S15–16). Discounting microminerals, the trend of elemental enrichments in generic flakes of CM is indistinguishable from that of the matrix, and thus very different in both magnitude and distribution to the enrichments measured in irregular clots and particle coatings.
Microstructure-Specific Trace Element Signatures
Clot and coating morphologies are characterised by similar negative δ13C values, albeit with slightly more negative values in coatings (centred on −15.16‰) than in clots (centred on −9.44‰). Flakes show a wide range of δ13C (between −47.4 ± 8.3‰ and +13.5 ± 2.0‰) including some positive values (Fig. 3). Clotted and particle coating morphologies are therefore characterised by restricted carbon isotope fractionations more indicative of biological processing.
Spatially correlated enrichments in trace elements, particularly transition metals, are found throughout the CM of irregular clots and particle coatings (Fig. 6a), but not in generic carbon flakes, where enrichments are localised within microminerals (Fig. S16). The average metal and metalloid enrichments within clotted and coating CM deviate notably from those in flakes and in the matrix, as evidenced by metal:metal ratios for V, Co, Ni and Cu against ubiquitously present bio-essential Fe (Fig. 6b; and compare average enrichments in Table S2 with those in Table S3). Since elemental enrichments in clots and coatings are not due to mineral phases, the diffuse trace element signature in both of these CM morphologies is primary and indigenous to CM. Moreover, since high-resolution optical microscopy was used to ensure that regions of interest were selected far from secondary veins or obvious deformational features (Fig. S9) through which later migration of fluids could have influenced this CM, and since hydrothermal silicification is extremely rapid (initiated in hours24,25,43) and provides a ‘time capsule’ of crystallographic preservation, post-diagenetic mobilisation of metals into or out of biological organic matter can be discounted. Furthermore, the rapid sealing of functional groups by silica during earliest diagenesis25 inhibits the uptake of metals from the silicifying fluid. Thus, since the CM is highly likely of biological origin (see Supplementary Discussion), we interpret trace element concentrations within this CM as reflecting the palaeo-metallome of the precursor biomass.

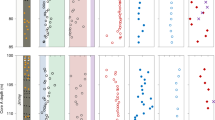
Systematic approach to calculating the palaeo-metallome with reference to modern organisms. (a) Comparison between fractional contributions of elements of interest in Josefsdal Chert carbonaceous material and reported prokaryotic metallomes (e.g. Zerkle et al.41). A metallomic contribution of V, Co, Ni, Cu and As (with bio-essential Fe, not shown) is frequently seen in irregular clots and particle coating morphologies. The least deviating pattern is seen between that of M. jannaschii and Josefadal Chert carbonaceous material: V, Ni, Co, Cu and Fe, together with reduced (or absent) requirements of Mn and Mo. The higher dependency on Cu seen for S. tepidum and S. meliloti more closely aligns its intracellular metal abundances with carbonaceous material in particle coatings, but we suggest that Cu may not be integral to the Josefsdal Chert carbonaceous material, but instead be the result of non-specific uptake due to extracellular polymer-driven resistance to toxicity (after Hickman-Lewis et al.33). In most cases, the concentrations in putative biomass is lower than that for in vivo cells. This is consistent with degradation of biomass during the decay of cellular components. (b) Metal:metal ratios for the studied microstructures against values in the matrix demonstrating statistically distinct enrichments of V, Co, Ni, Cu, K and Ca. Enrichments in transition metals and depletions in Ca and K are consistent with bio-accumulation (i.e., in molecular nanomachines) and toxicity mitigation (i.e., against salt stress). The overall differences between significant metal enrichments in clots and coatings (highlighted in green) with respect to flakes and the matrix (highlighted in grey) implies that the accumulation of transition metals in flakes occurs by a different, less efficient process. We propose bio-accumulation for the effective accumulation of metals in clots and coatings, but passive, non-specific accumulation in the case of flakes. Passive accumulation is consistent with the similarity in metal:metal ratios in flakes and the matrix. Where no box plot is shown, either one or both of the elements in question were not present in the region of interest.
Caution is yet required in the interpretation of this signature, since it may reflect a combination of original cellular chemical selection19,61, very limited chelation of elements during diagenesis24,25 and the uptake of elements into extracellular polymers as a response to toxicity33. Indeed, cellular and secreted biological molecules are commonly rich in carboxylate groups, peptidoglycan, teichuronic acid and other heteropolymeric polysaccharides, and are likely to retain metals accumulated during life even after death24,25,62. An understanding of the biogeochemistry of each element is thus required in order to decode the palaeo-metallome: a challenging, but potentially powerful, bio-indicator.
Origin of the Palaeo-Metallomic Biosignature
Metals are essential to all life and have widespread utility in molecular nanomachines19,20,21,23. Of the genome, proteome and metallome, the metallome is the only likely candidate for preservation throughout deep time. Indeed, biomarkers indicative of the proteome are not thought to preserve into Archaean sediments42. The spatially discontinuous metal-rich signatures observed in our analyses (Figs. 4–5) are a function of micrometric discontinuity in CM, which occurs as dispersed, interleaved fragments between silica crystals (Fig. 2g,h). This distribution is consistent with that of originally cellular material in similarly ancient and younger Proterozoic rocks59,63, i.e., micro-scale cross-sectional morphology bears little significance on the biological origin of CM. Generally, µPIXE mapped higher concentrations of elements in regions of visually elevated CM concentrations, however, the patterns of distribution of metals are frequently heterogeneous (Fig. 4c), which may reflect multiple micro-scale sources, i.e., precursor molecular diversity. Although some elements were present below the theoretical detection limit of PIXE (~100 ppm for most elements), the ability to map their presence in discrete energy channels is consistent with their occurrence at low concentrations56.
In hydrothermally influenced environments and potentially the wider Archaean ocean, soluble FeII, NiII, CoII and MnII were more bio-available, whereas insoluble CuII and ZnII and MoIV were less so61,64. The concentrations of these elements at the ocean-sediment interface would have been elevated by their incorporation into fluids permeating mafic oceanic bedrock. Our measured concentrations corroborate this: in the ambient environment, Cu, Zn and Mo are present in significantly lower concentrations than Fe, Ni and Co. Mo, Cu and Zn were, indeed, seemingly later (syn-GOE) additions to the metallome61,65, their importance to biology significantly post-dating the deposition of these rocks. Phylogenomic analyses of Archaean-representative prokaryotes suggest that their metal requirements were consistent with a ferro-sulphidic environment, for instance the necessity for Co66,67 and a lacking requirement of Zn64, high Cu sensitivity and low metal tolerance in the absence of ferro-sulphidic conditions20,64. These expectations are also supported by our results; indeed, almost all expected metallic elements are enriched in both CM and the matrix relative to their concentrations in the modern oceans. The Archaean CM studied further concentrates only a specific range of trace elements relevant to prokaryotic metallomes, including Fe, V, Co, Ni and As29,30,41,61, sustaining our hypothesis that clear and recurrent patterns of elemental enrichment in CM reflect the exceptionally preserved metallomic complement of biomass.
These enrichments are further consistent with coordination chemistry involving metalloproteins yielding ion enrichments within biomass23,68. Hard and soft Lewis acid and base (HSAB) theory predicts that hard Lewis acids (K, Ti, Fe, V, Cr, Mo, Mn) and soft Lewis acids (Ni, Co, Cu), together with hard Lewis bases, e.g., P, associated with PO43− and H2PO4−, and S, a hard base in SO42− and soft base in ROH, R2S and S2O32−, would be associated with ligands including H2O, ROH, RCO2−, CO32−, NO3−, PO43−, SO42− and NH3, which complex readily in hydrothermal effluent to make elements bioavailable (although SO42− and NO3− are generally present at low concentrations in hydrothermal fluids). Coordination complexes formed from hard acids require hard bases. The presence of either carboxylate groups or phosphates is thus useful, e.g. cell wall proteins and EPS in biomass69. Moreover, microbial matter is generally extremely effective in the role of metal accumulation based on the fact that biomass (particularly membrane-forming polymers) has an exceptionally high surface area to volume ratio for interaction with metal ions in the immediate environment coupled with a net negative charge due to a preponderance of carboxylate and other lipopolysaccharides, together with peptidoglycan. In both Gram-positive and Gram-negative microbes, this yields an unmatched ability to accumulate metal ions24, which have been experimentally demonstrated to be retained after death25. The sealing of surficial functional groups by silica during diagenesis24,25 also inhibits metal uptake from the silicifying fluid. Coordination with aliphatic compounds or highly cyclised abiotic carbon produced by Fischer Tropsch-type and other wet hydrothermal reactions is, from the point of view of complexation, distinctly less favourable66. CM enriched in both hard and soft metals is thus consistent with biologically driven accumulation. This is further consistent with the lack of diffuse elemental enrichments within the negative control – generic CM particles – which instead simply sequester elements in matrix concentrations (Fig. 6b).
Using metal: metal ratios (V/Fe, Co/Fe, Ni/Fe and Cu/Fe, K/Fe and Ca/Fe), we find that strikingly distinct trends emerge between the ratios in clots and coatings when compared with the ratios in flakes and the matrix (Fig. 6b). Clots and coatings generally show higher ratios of V/Fe, Co/Fe and Ni/Fe (as well as Cu/Fe, but see below) that suggest specific directional enrichment reflecting a controlled environment (cf.22). Flakes have metal:metal ratios closer to matrix values, suggesting a predominance of passive accumulation. Furthermore, the lower K/Fe and Ca/Fe ratios in clots and coatings than in flakes and the matrix further distinguish these two groups of microstructures. These recurrent patterns of metal incorporation argue for selective incorporation, occurring before post-mortem diagenesis. Diverse spatial concentrations of elements on the micron-scale also suggest slightly different concentration methods in the biogeochemical cycle of each element, whether for metabolism or detoxification.
We thus find that the elemental enrichments reported result from syngenetic environmental-organismal effects (i.e., metallomic compositions related to biomolecules), since diagenetic and taphonomic histories related to secondary alteration can be excluded. CM in both clots and coatings likely consists of intermixed cellular material reflecting a metabolic network or biocoenosis14,31, together with extracellular biomolecules and metal-chelating proteins, both of which contain anions (cell proteins and aspartic and glutamic acids) able to chelate metal cations, for example Cu33,69. Significant metal accumulation associated with potential siderophores and other highly chelating proteins70 not of direct relevance to the metallome would have been removed with minerals (i.e., point sources of extreme enrichment; Figs. 2e,f, S4–S8) during spatial quantification (see methods described in Supplementary Material) and would not hinder the distinction of metal accumulation within CM itself. As noted by Heim et al.58 it is difficult, and indeed often impossible, to distinguish microbially and abiotically mediated metal accumulation in modern deposits. Consequently, we advocate that inferences of metal accumulation ascribed to microbial activity should only be made based on elemental signatures in CM itself.
Metabolism of the Precursor Biomass
In the absence of cellular preservation, Linnaean classification is impossible, thus the elemental composition must be conservatively interpreted to represent a community of organisms. This makes comparison with previously reported single organism metallomes (e.g.29,30,41) challenging, and suggests that a more appropriate comparison would be with metabolism-specific metallomes30,41. δ13C values of this kerogen are consistent with multiple metabolisms known or hypothesised to have developed by the Palaeoarchaean, including anaerobic photosynthesis, sulphur and sulphate reduction, nitrogen fixation and methanogenesis (see Fig. 3), each of which may have existed in this microbial biome. Although many metabolisms depend upon a range of enzymatic co-factors23, certain metabolisms show specific requirements: hyperthermophile methanogens such as Methanocaldococcus jannaschii exhibit elevated metallomic fractional requirements of Ni, Co, Cu and V relative to other metabolisms, whereas Zn and Mn are required at substantially reduced concentrations22,40. Until Mo became more bioavailable at 2.5 Ga, nitrogen fixation is thought to have relied upon Fe and V71. Nonetheless, off-axis hydrothermal systems are known to mobilise small quantities of Mo72 and the anoxic weathering of volcanics could have sustained nitrogen fixation using Mo-nitrogenase at even 3.2 Ga73. Consequently, the lack of detection of appreciable concentrations of Mo in JC CM may simply reflect an absence of Mo-based nitrogen cycling in this environmental setting. Differential requirements for elements, although complex and highly dependent upon their palaeoenvironmental context, could conceivably define a biosignature for contributions by specific metabolisms in the precursor biomass and explain, as an example, the high concentrations of V retained within the studied kerogen. In Fig. 6a, we report µPIXE-determined spatially quantified elemental concentrations as percentage contributions to metallomes following the approach of Zerkle et al.41. Strong similarities with the fractional requirements of certain trace elements in specific metabolic machinery emerge.
Fe, Na, Mg, P, S, K and Ca are bio-essential41,61,65,74 and, with the exception of Ca, are distributed throughout the clotted and coating-like CM studied herein. This exceptional elemental complement alone is strong evidence for this CM having a biogenic origin. V is considered either bio-essential or bio-functional40,70 and has high retention potential within CM62. Very high fractional contributions of V in clots and coatings may stem from this preferential retention or original enrichment and, while potentially an overestimate relative to enrichments in other elements, suggests metabolic contributions from methanotrophic and diazotrophic organisms23,41. V is essentially absent in the matrix; this can be explained by its concentration within CM which, while abundant in the regions of interest, is negligible in bulk sample (<10 ppm; Fig. S11). Cu appears to have been incorporated into the metallome only from the Mesoarchaean onward61,65, thus its enrichment in CM probably reflects non-specific sequestration by biomolecules due to toxicity, as observed in polysaccharides associated with modern microbes33. Indeed, the elevated Cu/Fe ratios observed in the studied CM microstructures may be explicable by passive absorption by EPS22,33. We do not suggest that Cu was integral to the metabolic network of this 3.33 Ga microbial community, since this is incompatible with most theories of evolutionary biology, and since REE+Y palaeoenvironmental reconstruction (Fig. 2a) does not suggest any aspect of an oxygen oasis in the JC. Co, Ni and As are considered either bio-essential or bio-functional and are enriched within individual sulphide grains and more diffusely within CM. The modelled metallomes of chemoautotrophic methanogens and, more broadly, (hyper)thermophiles, demonstrate an enrichment of Ni to a fractional contribution twice that of other metabolisms30,41. Co and Ni are furthermore important components of the metallomes of methane- and nitrogen-cycling chemolithotrophs75, which are plausible metabolic precursors in light of the petographic context of this carbonaceous material: lithotrophs would be most likely associated with particles31. Such primitive, anaerobic metabolisms35 are also highly compatible with the thermophilic hydrothermal biomes evidenced by our trace and REE + Y environmental reconstruction (Figs. 2a, S10–S11) and thus the measured pattern of enriched Fe, Ni, Co, V and As is concordant with the expected metabolic network in this hydrothermal-lithotrophic palaeoenvironment. Of these elements, As is the most challenging case to evaluate since, although As-based metabolisms have been inferred at 2.7 Ga22, As is not widely used in metalloenzymes. We propose that its accumulation in these CM microstructures may either result from biological use or non-specific detoxification uptake as is potentially the case for Cu (e.g.33). Other bio-functional elements, including Ba, W, Cd and Sn, were not detected during our analyses. These elements are either absent from the palaeo-metallome of any biomass analysed or present in vanishingly small concentrations below the limit of detection. This discussion highlights the importance of evaluating a putative palaeo-metallome in light of the biogeochemistry of each of its constituent elements.
In conclusion, a thermophilic, hydrothermally influenced biome in the Palaeoarchaean would demand a metallome consisting of bio-essential elements together with metabolism-specific V, Cr, Co, Ni, As and W, and possibly others61,74. Due to bio-availability constraints in anoxic environments, Zn, Cu and Mo likely played a minor metallomic role on the Earth prior to the GOE23,61. Internally heat-driven, anaerobic conditions were plausibly more clement to thermophiles demanding metallomic V, Co, Ni, As and W, which is sustained by trophic-level comparisons with reported metallomes29,30,41. Our results are further consistent with expected elevated ion concentrations in the Archaean oceans21,64. We suggest that the trace element enrichments characterising both irregular clots and particle coatings in the 3.33 Ga Josefsdal Chert represent bioaccumulation (after27,28,68) in a hydrothermally influenced shallow-water biome. The trace element signature of this CM carries a strong resemblance to anaerobic, methanogenic or diazotrophic, thermophiles (and likely a combination of the two, and potentially other metabolisms such as sulphate reduction) in terms of both absolute and relative enrichments and fractional contributions to the palaeo-metallome (Fig. 6a). Moreover, metal:metal ratios show clear distinctions between the elemental compositions of irregular clots and particle coatings versus flakes and the matrix. The differences in these ratios are incompatible with passive accumulation, since all CM morphologies were deposited contemporaneously. In situ carbon isotope fractionations corroborate our biological interpretation of clots and coatings: values centred around values less negative than −20‰ are consistent with fractionation by nitrogen-cycling, sulphur-cycling, methanogenic and/or anoxygenic phototrophic microbes, although slightly heavy to infer methanotrophy9,15,16,17,76,77. We therefore propose that clotted and coating morphologies of CM are relics of an early Earth chemosynthetic (potentially chemolithotrophic) ecosystem.
The wide range of isotope fractionation in flakes, encompassing both positive and negative values, does not resemble known biological distributions16,17, but does exhibit a certain similarity to hydrothermal carbonaceous products53. Whether this indicates that the flakes and other generic CM analysed derive from abiotic sources alone is difficult to assess from solely carbon isotopes, significant though such a statement would be in such ancient material. Answering such a question would likely require high-resolution structural and geochemical analyses on micro-sampled material from multiple CM morphologies (see procedures described in78). Consequently, this dataset alone implies that these flakes resulted from diverse, non-specific processes.
Implications for the Metabolic Networks of Early Ecosystems
Elements must be bio-available in their ambient chemical environment to become an integral part of the metallome. These results provide the first estimation of the trace element budget of Archaean biomes (see also79,80), and support that Palaeoarchaean hydrothermally influenced habitats were rich in many of the elements required by the thermophilic prokaryotic metallome. Not all bio-functional metallic elements are present (e.g. W and Sn), and thus the metallome may have undergone degradation as a function of cell death and taphonomy, but it is more likely that these elements simply played no role in metabolisms of the precursor biomass since preservation in fossiliferous chert Lagerstätten is otherwise exceptional5,9,37,38. Palaeo-metallomic biosignatures are likely a function of both original cellular concentrations and the preservation potential of the intra- and extra-cellular components with which the element was associated (i.e., metalloproteins versus cytoplasm versus EPS). In Archaean sediments, the palaeo-metallome is most accurately assessed as a community signal. The approach pioneered herein opens new avenues of life detection in geological materials of any age and an opportunity to re-interrogate contentious putative biosignatures. Most significantly, it provides a means of investigating biogenicity in the absence of cellular preservation. In the near future, the co-occurrence of trace elements in possible CM will be measurable during Mars rover missions, and this type of biosignature could be observed with the PIXL instrument aboard the Mars 2020 rover81. Since Mars rovers lack the microscopy capabilities to observe individual microfossils, elemental signatures associated with putative biomass should be considered highly strategic biosignature targets.
In terms of the early terrestrial biosphere, our findings empirically support that the hypothesis of Frausto da Silva and Williams20 – that modern biological dependency on trace elements was influenced by the environments in which early life evolved – is correct. On a regionally thermophile early Earth, hydrothermally influenced oceans were environments relevant to the evolution of the prokaryotic metallome. Since the early aeons accounted for a large portion of microbial evolutionary history3,37,82, it is perhaps unsurprising that these elemental legacies have endured.
Methods
Sample collection
Samples were collected during field campaigns to the Barberton greenstone belt between 1999 and 2014, during which time a detailed appraisal of the stratigraphy and sedimentology has been conducted5,31,32,38. The studied samples were taken from the Josefsdal Chert between Ekulindeni, Mpumalanga province, South Africa, and Bulembu, Hhohho region, Eswatini. Three major sets of outcrops, from which the samples studied herein were obtained, are located at: 25°57.945′N, 31°04.720′E; 25°57.945′N, 31°04.798′E; 25°57.941′N, 31°04.722′E.
Fundamental petrographic characterisation
For optical microscopy, an Olympus BX51 microscope (CNRS-CBM, Orléans) was used. Unless stated otherwise, all optical images are in plane polarised light. Raman spectroscopy used a WITec Alpha500 RA (CBM, Orléans) equipped with a green laser at 532 nm wavelength and a laser power of 5 mW to avoid thermal alteration of the sample. Raman geothermometry used the carbonaceous materials-based geothermometer of Kouketsu et al.54. Qualitative identification of spectral characteristics suggested our materials reflect medium metamorphic grade (i.e., lower greenschist facies), after which we used Equation 2 of54) as follows: T(°C) =−6.78(FWHM–D2) + 535, where FWHM is the full width half maximum and D2 is a band in the first-order disordered region (i.e., ~1200–1700 cm−1). SEM analyses used Hitachi T3000 (ITSO, Orléans) and JEOL (Dipartimento BiGeA, Università of Bologna) instruments operating at 5 keV and 15 keV. Elemental maps were run for 150–200 frames in order to accrue sufficient counts, and point analyses for 100–300 s for the same reason.
Particle-induced X-ray emission spectroscopy
Particle-induced X-ray emission (µPIXE) combined with Rutherford Backscattering Spectrometry (RBS) was performed using the microbeam beamline of the AIFIRA facility (CENBG, Gradignan), which is described in detail in Sorieul et al.55. This used the 3 MeV proton microbeam (1 µm) with a current of 200 pA, and a high energy range of 40 keV to mitigate the problem of overlap amongst K and L X-ray lines57. Three Si-detectors (one RBS and two PIXE) collected data: the RBS was used for charge monitoring, as lithological samples are insulators, whereas the two PIXE detectors were used for elemental quantification and mapping. The first PIXE detector was equipped with an Al-“Funny Filter” (thickness 100 µm, hole size 2 mm) and a Kapton filter (thickness 50 µm). Kapton is used to filter out elements at lower atomic weight, and is therefore key to determining the true concentrations of heavier trace elements, particularly for those of Ti and higher atomic mass. Greater hole size increases count rate, but has no effect on the rate of individual element detection. The second PIXE detector was equipped with an Al-“Funny Filter” (thickness 100 µm, hole size 1 mm) and was used for complete characterisation of the sample. No filter was used for the RBS detector. Each analysis was allowed to run for 8–12 hours. All analyses were conducted with a dead time of less than 10% in order to avoid pile-up and spectra distortion. Dependent upon the size of the region of interest, scans are either 50 µm, 100 µm or 200 µm squares. PIXE is a non-destructive, non-invasive, in situ method for measuring trace element data in cherts when compared with efforts involving HF total digestion. Multiple examples of each morphology of carbonaceous matter have been measured to produce a statistically significant dataset, depending upon their abundance in the studied samples. The matrix invariably contains a small amount of disaggregated carbonaceous matter of indeterminate origin, thus serves as the ideal benchmark against which elemental concentrations of specific larger microstructures are normalised (i.e. ambient conditions). For each sample, multiple analyses of the matrix adjacent to microstructures of interest were conducted, providing the average matrix value against which analyses of carbonaceous material within that microstructure were normalised. X-ray counts can be converted into concentrations following the rationale and methods described in Halden et al.56 and Campbell et al.57 which detail the utility and capability of PIXE of determining spatially resolved concentrations of the elements of interest at micrometric resolutions.
The quantification of PIXE data involves a sequential workflow using three programs: SupaVisio, SIMNRA and Gupix57,83,84. SupaVisio permits the selection of regions of interest, the spectra of which are extracted from the spectrum of the entire region of analysis. Each extracted RBS spectrum was treated with SIMNRA for charge determination84. The charges determined were used for the quantification of sixteen elements of interest (P, S, K, Ca, Ti, V, Cr, Mn, Fe, Co, Ni, Cu, Zn, As, Zr, Mo) with Gupix83. The algorithm used by Gupix83 calculates a limit of detection for the alpha- and beta-ray (K) of each element (quantification of L and M rays is also possible, though was not of relevance to the elements in question), from which the user assesses the presence or absence of the element. Where close to the limit of detection (within 10% considering analytical error), we took a conservative approach and deemed the element absent. Therefore, all stated concentrations are low estimates. In all quantifications, we consider a ‘mineral effect’. This was identified based upon the element maps generated in SupaVisio, since while many trace elements are broadly enriched within carbonaceous matter, they are occasionally especially enriched within small regions (i.e. minerals). Discounting any quantified analysis in which the value determined is more than five times the average of all other analyses of that type removes the ‘mineral effect’ for that element. These regions were often found to correlate with optically identifiable mineral phases. The application of the ‘mineral effect’ may prevent the quantification of concentration within an element at depth where it is not directly identifiable by petrographic observation but nonetheless skews the results to higher averages (pre-screening of the regions of interest was conducted using high-resolution optical microscopy before µPIXE). This has been done on an element-by-element basis such that only one datapoint will be removed from the overall quantification, thus maintaining the integrity of the statistic.
There is no direct correlation between the appearance of concentrations in the maps, which are a function of the number of counts at any specific point, and the concentration (ppm) of that element, which is a function of the detector-specific quantification of that element according to the algorithm of Gupix (see56,57,83). Although the detection of some elements is below the theoretical detection limit of PIXE (~100 ppm for most elements), the ability to map their occurrence in discrete energy channels is consistent with their occurrence as shown in the individual element maps, but at concentrations well below the theoretical limit of detection for PIXE (<100 ppm)56,57. The limit of detection (LOD) is defined as the concentration of an element that would give rise to an X-ray peak having an intensity equal to the three-sigma fluctuation of the background underlying the peak. In GUPIX software, LOD is defined as 3 times the square root of the background over 1 full-width half-maximum centred on the principal peak centroid. This corresponds to the LOD definition used for fluorescence spectrometry. The value of LOD can vary with the concentration of the detected element, the preparation of the sample and more generally with the conditions of analysis. In order to achieve both the best conditions of analysis and the lowest background, we used the soft borosilicate glass NIST1411 standard as a reference material. We also prepared the samples accordingly to avoid any parasitic radiation due to the interaction of the beam with the chert matrix. PIXE standardisation is based on the accurate determination of an instrumental constant independent of the X-ray energy. In practice, several uncertainties arose mainly from the inaccurate characterisation by the detector and uncertainties in X-ray transmission. GUPIX software takes into consideration all of these deviations from the ideal measurement through the determination of a sole parameter termed the “H-factor”. For that purpose, two standards were used: soft borosilicate glass NIST1411 and stainless steel AISI 15-7PH. The combination of these two standards covers the energy range from 1 to 20 keV in which all the potential trace elements could be detected. NIST1411 was also used to evaluate the matrix effect and finely tune the beam conditions and detection set-up for the analysis of these cherts.
Carbon isotope ratio mass spectrometry
In situ carbon isotope ratio determinations were performed using a CAMECA IMS1280 large-geometry SIMS instrument at the Swedish Museum of Natural History operating in scanning ion imaging mode. Following a 60 sec. pre-sputter with a 20 kV incident energy, ca. 1 nA Cs beam rastered over a 25 ×25 µm area to remove the gold coating, the beam was reduced to a critically focussed <1 µm analytical spot, with a beam current of ca. 100 pA, which was rastered over an area of 20 × 20 µm during acquisition of data. A low energy electron flooding gun was utilised to prevent charge build up on largely insulating target areas. Sputter secondary ions were steered back onto the ion optic axis using the dynamic transfer optical system, a secondary raster that is synchronised with the primary raster to permit acquisition of data from a scanned area at high mass resolution. The secondary ion species 12C- and 13C- were separated at a mass resolution of 4000 (M/ΔM), in order to resolve 13C from 12C1H and detected sequentially in the axial ion-counting electron multiplier (EMax) with counting times of 1 sec. and 5 sec. respectively over 100 cycles. The resulting ion images were processed using CAMECA Winimage2 software to identify regions of interest (ROIs) of high C concentration, from which the 13 C/12 C ratios were calculated, with the 44 ns EMax deadtime correction applied at the pixel level. Analyses of an in-house reference material, Cpyr2, a pyrolised graphite disk with a δ13CPDB value of −35.4‰ (G.D. Layne, Memorial University of Newfoundland, pers. comm.) were performed using exactly the same primary beam and raster conditions in order to retain identical sputtering conditions and, hence, identical mass bias relative to the target graphitised carbonaceous material. Nonetheless, the much higher secondary count rates from a 100% graphite target precluded use of the EM and instead data were acquired simultaneously in two low noise Faraday Cups (FCs), 13C in the axial FC2 and 12 C in L’2 FC operating at mass resolutions of 4000 and 2530 respectively. The instrumental mass bias determined from the Cpyr2 analyses was then applied to the ratios obtains from the target ROIs to yield their δ13CPDB. Some of the near-positive and positive values were derived from high isotopic values located at the very edge of the rastered area and could result from a bias in beam centring, which may generate a concomitant bias in isotopic values.
Inductively coupled plasma mass spectrometry
Fresh rock chips, unaffected by secondary alteration and veining, were selected and ground to a particle size of less than 4 µm for ICP-MS analysis. ICP-MS analyses were conducted at the Centre de Recherches Pétrographiques et Géochimiques (CRPG), Nancy, France, using Thermo Fisher ICap 6500 and Agilent 7700X instruments.
References
Walsh, M. M. & Lowe, D. R. Modes of accumulation of carbonaceous matter in the early Archaean: a petrographic and geochemical study of the carbonaceous cherts of the Swaziland Supergroup. In: Lowe, D. R., Byerly, G. R. (Eds.) Geologic Evolution of the Barberton Greenstone Belt, South Africa. Geological Society of America Special Paper 329 Boulder, Colorado, pp. 115–132 (1999).
van Zuilen, M. A., Chaussidon, M., Rollion-Bard, C. & Marty, B. Carbonaceous cherts of the Barberton greenstone belt, South Africa: isotopic, chemical and structural characteristics of individual microstructures. Geochimica et Cosmochimica Acta 71, 655–669 (2007).
Brasier, M. D., Wacey, D., McLoughlin, N. Taphonomy in temporally unique settings: an environmental traverse in search of the earliest life on Earth. In Allison, P. A. & Bottjer, D. J. (Eds.), Taphonomy: Process and Bias Through Time, Topics in Geobiology 32, Springer (2011).
van Zuilen, M. A. The significance of carbonaceous matter to understanding life processes on early Earth. In Van Kranendonk, M. J., Bennett, V. C., Hofmann, J. E. (Eds.) Earth’s Oldest Rocks, Elsevier, pp. 945–963 (2018).
Westall, F. et al. Implications of in situ calcification for photosynthesis in a ∼3.3 Ga-old microbial biofilm from the Barberton Greenstone Belt, South Africa. Earth and Planetary Science Letters 310, 468–479 (2011).
Gourier, D. et al. Extraterrestrial organic matter preserved in 3.33 Ga sediments from Barberton, South Africa. Geochimica et Cosmochimica Acta 258, 207–225.
Javaux, E. J. Challenges in evidencing the earliest traces of life. Nature 572, 451–460 (2019).
Marshall, C. P. et al. Structural characterization of kerogen in 3.4 Ga Archaean cherts from the Pilbara Craton, Western Australia. Precambrian Research 155, 1–23 (2007).
Lepot, K. et al. Texture-specific isotopic compositions in 3.4 Gyr old organic matter support selective preservation in cell-like structures. Geochimica et Cosmochimica Acta 112, 66–86 (2013).
Greco, F., Cavalazzi, B., Hofmann, A., Hickman-Lewis, K. Raman spectroscopy in palaeontology: a case study from Archaean biostructures. Bollettino della Società Paleontologica Italiana 57, 59–74.
Rosing, M. T. 13C-depleted carbon microparticles in>3700-Ma sea-floor sedimentary rocks from west Greenland. Science 283, 674–676 (1999).
Grassineau, N. V., Appel, P. W. U., Fowler, C. M. R. & Nisbet, E. G. Distinguishing biological from hydrothermal signatures via sulphur and carbon isotopes in Archaean mineralizations at 3.8 and 2.7 Ga. In McDonald, I., Boyce, A. J., Butler, I. B., Herrington, R. J., Polya, D. A. (Eds.) Mineral Deposits and Earth Evolution, Geological Society of London Special Publications 248, 195–212 (2005).
Duda, J.-P. et al. Ideas and perspectives: hydrothermally driven redistribution and sequestration of early Archean biomass – the “hydrothermal pump hypothesis”. Biogeosciences 15, 1535–1548 (2018).
Schopf, J. W. et al. An anaerobic ~3400 Ma shallow-water microbial consortium: Presumptive evidence of Earth’s Paleoarchean anoxic atmosphere. Precambrian Research 299, 309–318 (2017).
Schidlowski, M. Carbon isotopes as biogeochemical recorders of life over 3.8 Ga of Earth history: evolution of a concept. Precambrian Research 106, 117–134 (2001).
Vieth, A. & Wilkes, H. Stable isotopes in understanding origin and degradation processes of petroleum. In Timmis, K. (Ed.). Handbook of Hydrocarbon and Lipid Microbiology, Springer, Heidelberg, pp. 97–111 (2009).
Havig, J. R., Hamilton, T. L., Bachan, A. & Kump, L. R. Sulfur and carbon isotopic evidence for metabolic pathway evolution and a four-stepped Earth system progression across the Archean and Paleoproterozoic. Earth-Science Reviews 174, 1–21 (2017).
McCollum, T. M. & Seewald, J. S. Carbon isotope composition of organic compounds produced by abiotic synthesis under hydrothermal conditions. Earth and Planetary Science Letters 243, 74–84 (2006).
Williams, R. J. P. Chemical selection of elements by cells. Coordination Chemistry Reviews 216–217, 583–595 (2001).
Frausto da Silva, J. J. R. & Williams, R. J. P. The biological chemistry of the elements. Oxford University Press, pp. 600 (2001).
Williams, R. J. P. & Fraústo da Silva, J. J. R. Evolution was chemically constrained. Journal of Theoretical Biology 220, 323–343 (2003).
Sforna, M. C. et al. Evidence for arsenic metabolism and cycling by organisms 2.7 billion years ago. Nature Geoscience 7, 811–815 (2016).
Moore, E. K., Jelen, B. I., Giovannelli, D., Raanan, H. & Falkowski, P. G. Metal availability and the expanding network of microbial metabolisms in the Archaean eon. Nature Geoscience 10, 629–636 (2017).
Schultze-Lam, S., Fortin, D., Davis, B. S. & Beveridge, T. J. Mineralization of bacterial surfaces. Chemical Geology 132, 171–181 (1993).
Orange, F., Disnar, J.-R., Westall, F., Prieur, D. & Baillif, P. Metal cation binding by the hyperthermophilic microorganism, Archaea Methanocaldococcus jannaschii, and its effects on silicification. Palaeontology 54, 953–964 (2011).
Lobinski, R., Becker, J. S., Haraguchi, H. & Sarkar, B. Metallomics: Guidelines for terminology and critical evaluation of analytical chemistry approaches (IUPAC Technical Report). Pure and Applied Chemistry 82, 493–504 (2010).
Wogelius, R.A. et al. Science 333, 1622–1626 (2011).
Edwards, N. P. et al. Leaf metallome preserved over 50 million years. Metallomics 6, 774–782 (2014).
Liermann, L. J., Hausrath, E. M., Anbar, A. D. & Brantley, S. L. Assimilatory and dissimilatory processes of microorganisms affecting metals in the environment. Journal of Analytical Atomic Spectrometry 22, 867–877 (2007).
Cameron, V., House, C. H. & Brantley, S. L. A first analysis of metallome biosignatures of hyperthermophilic archaea. Archaea 2012 Article ID 789278 (2012).
Westall, F. et al. Archean (3.33 Ga) microbe-sediment systems were diverse and flourished in a hydrothermal context. Geology 43, 615–618 (2015).
Westall, F. et al. A hydrothermal-sedimentary context for the origin for life. Astrobiology 18, 259–293 (2018).
Hickman-Lewis, K. et al. Mechanistic morphogenesis of organo-sedimentary structures growing under geochemically stressed conditions: keystone to the interpretation of some Archaean stromatolites? Geosciences 9, 359 (2019).
Hofmann, A. & Bolhar, R. The origin of carbonaceous cherts in the Barberton Greenstone Belt and their significance for the study of early life in mid-Archaean rocks. Astrobiology 7, 355–388 (2007).
Nisbet, E. G. & Fowler, C. M. R. The hydrothermal imprint on life: did heat-shock proteins, metalloproteins and photosynthesis begin around hydrothermal vents? Geological Society of London Special Publications 118, 239–251 (1996).
Gaucher, E. A. J. T., Kratzer, J. T. & Randall, R. N. Deep Phylogeny—How a Tree Can Help Characterize Early Life on Earth. Cold Spring Harbor Perspectives Biology 2, a002238 (2010).
Nisbet, E. G. & Fowler, C. M. R. Archaean metabolic evolution of microbial mats. Proceedings of the Royal Society of London B 266, 2375–2382 (1999).
Hickman-Lewis, K., Westall, F. & Cavalazzi, B. Trace of Early Life in the Barberton Greenstone Belt. In Earth’s Oldest Rocks, Second Edition (Eds. Van Kranendonk, M. J., Bennett, V. C. & Hofmann, J. E.), pp. 1029–1058 (2018).
Sleep, N. H. Geological and geochemical constraints on the origin and evolution of life. Astrobiology 18, 1199–1219 (2018).
Arndt, N. T. & Nisbet, E. G. Processes on the young Earth and the habitats of early life. Annual Review of Earth and Planetary Sciences 40, 521–549 (2012).
Zerkle, A. L., House, C. H. & Brantley, S. L. Biogeochemical signatures through time as inferred from whole microbial genomes. American Journal of Science 305, 467–502 (2005).
French, K. L. et al. Reappraisal of hydrocarbon biomarkers in Archean rocks. Proceedings of the National Academy of Sciences USA 112, 5915–5920 (2015).
Schultze-Lam, S., Ferris, F. G., Konhauser, K. & Wiese, R. G. In situ silicification of an Icelandic hot spring microbial mat: implications for microfossil formation. Canadian Journal of Earth Sciences 32, 2021–2026 (1995).
Gourcerol, B., Thurston, P. C., Kontak, D. J. & Côté-Mantha, O. Interpretations and implications of LA ICP-MS analysis of chert for the origin of geochemical signatures in banded iron formations (BIFs) from the Meadowbank gold deposit, Western Churchill Province, Nunavut. Chemical Geology 410, 89–107 (2015).
Bolhar, R., Hofmann, A., Siahi, M., Feng, Y.-X. & Delvigne, C. A trace element and Pb isotopic investigation into the provenance and deposition of stromatolitic carbonates, ironstones and associated shales of the ~3.0 Ga Pongola Supergroup, Kaapvaal Craton. Geochimica et Cosmochimica Acta 158, 57–78 (2015).
Jeandel, C. & Oelkers, E. H. The influence of terrigenous particulate material dissolution on ocean chemistry and global element cycles. Chemical Geology 395, 50–66 (2015).
Walsh, M. M. Microfossils and possible microfossils from the Early Archean Onverwacht Group, Barberton Mountain Land, South Africa. Precambrian Research 54, 271–293 (1992).
Margulis, L., Grosovsky, B. D. D., Stolz, J. F., Gong-Collins, E. J. & Lenk, S. Distinctive microbial structures and the pre-Phanerozoic fossil record. Precambrian Research 20, 443–477 (1983).
Juniper, S. K., Martineu, P., Sarrazin, J. & Gélinas, Y. Microbial-mineral floc associated with nascent hydrothermal activity on CoAxial Segment, Juan de Fuca Ridge. Geophysical Research Letters 22, 179–182 (1995).
Wacey, D., Saunders, M., Kong, C., Brasier, A. T. & Brasier, M. D. 3.46 Ga Apex chert ‘microfossils’ reinterpreted as mineral artefacts produced during phyllosilicate exfoliation. Gondwana Research 36, 296–313 (2016).
Rasmussen, B. & Buick, R. Oily old ores: evidence for hydrothermal petroleum generation in an Archean volcanogenic massive sulfide deposit. Geology 28, 731–734 (2000).
Wacey, D., Fisk, M., Saunders, M., Eiloart, K. & Kong, C. Critical testing of potential cellular structures within microtubes in 145 Ma volcanic glass from the Argo Abyssal Plain. Chemical Geology 466, 575–587 (2017).
Proskurowski, G. Abiogenic hydrocarbon production at the geosphere-biosphere interface via serpentinization reactions. In Timmis, K. (Ed.). Handbook of Hydrocarbon and Lipid Microbiology, Springer, Heidelberg, pp. 215–231 (2009).
Kouketsu, Y. et al. A new approach to develop the Raman carbonaceous material geothermometer for low-grade metamorphism using peak width. Island Arc 23, 33–50 (2014).
Sorieul, S., Alfaurt, Ph, Daudin, L., Serani, L. & Moretto, Ph Aifira: an ion beam facility for multidisciplinary research. Nuclear Instruments and Methods in Physics Research Section B: Beam Interactions with Material and Atoms 332, 68–73 (2014).
Halden, N. M., Campbell, J. L. & Teesdale, W. J. PIXE microanalysis in mineralogy and petrology. Canadian Mineralogist 33, 293–302 (1995).
Campbell, J. L., Teesdale, W. J. & Halden, N. M. Theory, practice and application of PIXE microanalysis and SPM element mapping. Canadian Mineralogist 33, 279–292 (1995).
Heim, C. et al. Assessing the utility of trace and rare earth elements as biosignatures in microbial iron oxyhydroxides. Frontiers in Earth Science 3, https://doi.org/10.3389/feart.2015.00006 (2015).
Wacey, D., Kilburn, M. R., Saunders, M., Cliff, J. & Brasier, M. D. Microfossils of sulphur-metabolizing cells in 3.4-billion-year-old rocks of Western Australia. Nature Geoscience 4, 698–702 (2011).
Marin-Carbonne, J. et al. Sulfur isotope’s signal of nanopyrites enclosed in 2.7 Ga stromatolitic organic remains reveal microbial sulfate reduction. Geobiology 2018, 1–18 (2018).
Rickaby, R. E. M. Goldilocks and the three inorganic equilibria: how Earth’s chemistry and life coevolve to be nearly in tune. Philosophical Transactions of the Royal Society A 373, 20140188 (2015).
Marshall, C. P. et al. Imaging of vanadium in microfossils: a new potential biosignature. Astrobiology 17, 1069–1076 (2017).
De Gregorio, B. T. & Sharp, T. G. The structure and distribution of carbon in the 3.5 Ga Apex chert: implications for the biogenicity of Earth’s oldest putative microfossils. American Mineralogist 91, 784–789 (2006).
Saito, M. A., Sigman, D. M. & Morel, F. M. M. The bioinorganic chemistry of the ancient ocean: the co-evolution of cyanobacterial metal requirements and biogeochemical cycles at the Archean-Proterozoic boundary. Inorganica Chimica Acta 356, 308–318 (2003).
Dupont, C. L., Yang, S., Palenik, B. & Bourne, P. E. Modern proteomes contain putative imprints of ancient shifts in trace metal geochemistry. Proceedings of the National Academy of Sciences of the United States of America 103, 17822–17827 (2006).
Moore, E. K. et al. Geological and Chemical Factors that Impacted the Biological Utilization of Cobalt in the Archean Eon. Journal of Geophysical Research: Biogeosciences 123, JG004067 (2018).
Saito, M. A., Moffett, J. W., Chisholm, S. W. & Waterbury, J. B. Cobalt limitation and uptake in Prochlorococcus. Limnology and Oceanography 47, 1629–1636 (2002).
Wang, S. et al. Variations in Metal Tolerance and Accumulation in Three Hydroponically Cultivated Varieties of Salix integra Treated with Lead. PLoS ONE 9, e108568 (2014).
de Oliveira Martins, P. S., de Almeida, N. F. & Leite, S. G. F. Application of a bacterial extracellular polymeric substance in heavy metal adsorption in a co-contaminated aqueous system. Brazilian Journal of Microbiology 39, 780–786 (2008).
Leventhal, G.E., Ackerman, M., Schiessl, K.T. Why microbes secrete molecules to modify their environment: the case of iron-chelating siderophores. Journal of the Royal Society Interface 16 (2019).
Boyd, E. S. & Peters, J. W. New insights into the evolutionary history of biological nitrogen fixation. Frontiers and Microbiology 4, 201 (2013).
Wheat, C. G., Mottl, M. J. & Rudnicki, M. Trace element and REE composition of a low-temperature ridge-flank hydrothermal spring. Geochimica et Cosmochimica Acta 66, 3693–3705 (2002).
Stüeken, E. E., Buick, R., Guy, B. M. & Koehler, M. C. Isotopic evidence for biological nitrogen fixation by molybdenum-nitrogenase from 3.2 Gyr. Nature 520, 666–669 (2015).
Robbins, L. J. et al. Trace elements at the intersection of marine biological and geochemical evolution. Earth-Science Reviews 163, 323–348 (2016).
Friedrich, B. & Schwartz, E. Molecular biology of hydrogen utilization in aerobic chemolithotrophs. Annual Review of Microbiology 47, 351–383 (1993).
Londry, K. L. & Des Marais, D. J. Stable carbon isotope fractionation by sulfate-reducing bacteria. Applied and Environmental Microbiology 69, 2942–2949 (2003).
McCandless, T. E. & Gurney, J. J. Diamond eclogites: comparison with carbonaceous chondrites, carbonaceous shales and microbial carbon-enriched MORB. Russian Geology and Geophysics 38, 394–404 (1997).
Cavalazzi, B. Chemotrophic Filamentous Microfossils from the Hollard Mound (Devonian, Morocco) as Investigated by Focused Ion Beam. Astrobiology 7, 402–415 (2007).
Hickman-Lewis, K. et al. Goldschmidt Abstracts 2017, 1627 (2017).
Hickman-Lewis, K. et al. Influence of early ocean chemistry on cell biochemistry and prokaryotic metallomic biosignatures. Goldschmidt Abstracts 2018, 1014 (2018).
Allwood, A. C. et al. Texture-specific elemental analysis of rocks and soils with PIXL: The Planetary Instrument for X-ray Lithochemistry on Mars 2020. IEEE Aerospace Conference Proceedings 2015, 15215581 (2015).
Knoll, A. H., Bergmann, K. D. & Strauss, J. V. Life: the first two billion years. Philosophical Transactions of the Royal Society B 371, 20150493 (2016).
Maxwell, J. A., Teesdale, W. J. & Campbell, J. L. The Gupix PIXE software package II. Nuclear Instruments and Method in Physics Research B 95, 407–421 (1995).
Mayer, M. SIMNRA, a simulation program for the analysis of NRA, RBS and ERDA. AIP Conference Proceedings 475, 541 (1999).
Acknowledgements
We are grateful to Giorgio Gasparotto (BiGeA-Bologna) for help with SEM work, and to Sylvain Janiec (ISTO-Orléans) for the production of thin sections. We acknowledge financial support from the French National Centre for Scientific Research (CNRS), the French Space Agency (CNES), the MASE (Mars Analogues for Space Exploration) project (EU-FP7 Grant no. 607297), and the INACMa (Inorganic Nanoparticles in Archaean Carbonaceous Matter - a key to early life and palaeoenvironmental reconstructions) project (EU-FP7 Grant no. 618657). The NordSIMS facility in Stockholm is supported by Swedish Research Council infrastructure grant no. 2017-00671 and the Swedish Museum of Natural History; this is NordSIMS publication no. 635. KHL received generous support from the COST ORIGINS Action in the form of Grant no. TD1308-37679, Grant no. TD1308–38914 and Grant no. TD1038–40795. We are further very grateful to Eva Stüeken and an anonymous reviewer for constructive comments and recommendations that significantly improved the manuscript.
Author information
Authors and Affiliations
Contributions
K.H.L. and F.W. conceptualised the study. B.C. and F.W. collected samples. K.H.L., S.S., P.G., F.F., M.J.W., H.J. and F.W. conducted the experiments. K.H.L., S.S., P.G., M.J.W. and H.J. analysed the data. K.H.L. devised and developed the idea of the palaeo-metallome. K.H.L. wrote the paper, with contributions from B.C., S.S., P.G., F.F. and F.W. K.H.L., B.C., S.S., P.G., F.F., M.J.W., H.J., T.G., C.S.C. and F.W. contributed to discussion of the data.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hickman-Lewis, K., Cavalazzi, B., Sorieul, S. et al. Metallomics in deep time and the influence of ocean chemistry on the metabolic landscapes of Earth’s earliest ecosystems. Sci Rep 10, 4965 (2020). https://doi.org/10.1038/s41598-020-61774-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-61774-w
This article is cited by
-
The Habitability of Venus
Space Science Reviews (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
